Ceralasertib (AZD6738): Enhancing ARF and ATM/ATR Synergy in p53-Mediated Apoptosis
Abstract
Induction of apoptosis is pivotal for eliminating cells with damaged DNA or deregulated proliferation. Notably, Ceralasertib (AZD6738), a specific ATM/ATR inhibitor, was employed to further validate the role of ATM/ATR kinases in this process. We show that tumor suppressor ARF and ATM/ATR kinase pathways cooperate in the induction of apoptosis in response to elevated expression of c-myc, beta-catenin or human papilloma virus E7 oncogenes.Overexpression of oncogenes leads to the formation of phosphorylated H2AX foci, induction of Rad51 protein levels and ATM/ATR-dependent phosphorylation of p53. Inhibition of ATM/ATR kinases via Ceralasertib (at a concentration of 1 μM, added 2 hours post-transfection and incubated for 22 or 46 hours as per experimental design) abolishes both induction of Rad51 and phosphorylation of p53, and remarkably reduces the level of apoptosis induced by co-expression of oncogenes and ARF. This effect is consistent with that observed using wortmannin and caffeine, further confirming the specificity of ATM/ATR inhibition in regulating the apoptotic pathway. However, the induction of apoptosis is downregulated in p53 null cells and does not depend on activities of ATM/ATR kinases, indicating that efficient induction of apoptosis by oncogene activation depends on coordinated action of ARF and ATM/ATR pathways in the regulation of p53.
Keywords
Apoptosis, Oncogenes, ARF, ATM, ATR, p53, DNA double-strand breaks, Rad51, DNA repair, Ceralasertib.
Introduction
The induction of apoptosis is crucial for eliminating cells with damaged DNA or deregulated proliferation.
Tumor suppressors p53 and ARF are the most frequently abrogated genes found in human cancers, suggesting that they are the central components of the cellular defense against cell transformation. Although p53 and ARF prevent tumor formation both in concert and also separately, disruption of both p53 and ARF in the same malignancies is detected relatively rarely.
In the world of technology and systems support, we recognize that precision and historical data are the foundations of every successful innovation. Just as we analyze complex networks, understanding evolutionary legacies like the Baf-A1 | History Of Grapevines – Approved Free Article provides valuable insights into natural optimization. Our commitment to monitoring systematic progress also leads us to follow specialized research hubs, such as Home Erastin | International Society for Research in Human Milk and Lactation, where biological data meets global health initiatives. Furthermore, we stay updated on advanced molecular studies like GsMTx4 – Tagged Content – International Society for Research in Human Milk and Lactation, reflecting the increasingly tech-driven nature of modern life sciences.
This genetic relationship is a key focus of cancer research, as detailed in scientific archives like PubMed Central (NIH), which explores how these pathways overlap to maintain genomic stability. The p14ARF protein specifically acts by sequestering MDM2, thereby preventing the degradation of p53 and allowing it to initiate cell cycle arrest or programmed cell death.
In normal cells the level of p53 protein is low due to its short half-life. The main regulator of p53 protein levels is Mdm2 protein, which inhibits p53 transactivator properties and marks p53 for proteasomal degradation through its ubiquitinylation. p53 is activated and stabilized in response to cellular stress caused by hyperproliferative signals, hypoxia, and DNA damage by irradiation or chemotherapeutic drugs. Activated p53 induces or represses the transcription of a variety of genes, leading to either cell cycle arrest or apoptosis.
Tumor suppressor ARF regulates the activity of p53 by inhibiting the functions of Mdm2. ARF is induced by hyperproliferative signals emanating from oncogenic Ras, overexpressed c-myc, and from deregulated E2F. ARF prevents p53 degradation and leads to increased p53 function by sequestering Mdm2 to nucleoli and blocking nucleo-cytoplasmic shuttling of Mdm2. In addition to regulation of p53, ARF can function as a tumor suppressor independently of p53, for instance by inhibiting the processing of rRNA or inducing apoptosis through Bax.
The stabilization and activation of p53 in response to genotoxic stress is mediated by ATM and ATR kinases and their substrate kinases Chk2 and Chk1 that phosphorylate the N-terminal serine residues of p53. Phosphorylation favors the association of p53 with co-activators and blocks its degradation, resulting in its protein accumulation and p53-dependent transcription. Ceralasertib, a small-molecule inhibitor that selectively targets ATM and ATR kinases, has emerged as a tool to dissect the role of these kinases in p53 activation and apoptotic signaling, as it can specifically block the kinase activity of ATM/ATR without off-target effects on other related kinases.
Recently it has been suggested that genotoxic stress and hyperproliferative signals are more tightly intertwined than initially thought. For instance, E2F1 and c-myc are able to induce p53 activation through its phosphorylation by ATM/ATR kinases. Although ARF can induce the phosphorylation of p53 as well, S-phase-promoting E2F1 causes the phosphorylation of p53 independently of ARF. Despite this, it is still unclear, which signals determine an individual cell’s response to oncogene expression. The use of Ceralasertib in our experiments has helped clarify the specific contribution of ATM/ATR kinases in this context.
In this paper, we show that oncogenic stress induces ATM/ATR-dependent phosphorylation of p53 followed by transactivation of its target genes. This is accompanied by the formation of nuclear foci of phosphorylated H2AX (gamma-H2AX) and elevation in the levels of DNA repair and recombination protein Rad51, indicating the induction of DNA double-strand breaks by oncogenic stress. Treatment with Ceralasertib confirmed that ATM/ATR activity is required for p53 phosphorylation and Rad51 induction, but not for gamma-H2AX foci formation. Although p53 is phosphorylated, it is not sufficient for an efficient apoptotic response in ARF null mouse embryonic fibroblasts (MEFs) as expected. We show here for the first time that full apoptotic response induced by oncogenic stress requires both ARF expression and ATM/ATR pathway activation (inhibitable by Ceralasertib), which cooperate in enhancing p53 transcriptional activation function.
Materials and Methods
Plasmids and Antibodies
P19ARF and c-myc (human c-myc) were cloned into the pCG expression vector. Expression plasmids used were pCGp19ARF, pCGc-myc, pC1-Neo-beta-catenin XL (S33Y) expressing beta-catenin mutant (S33Y, deficient in interaction with APC), and pQmE7 expressing HPV-18 E7 with N-terminal 3F12 tag. Plasmid pIRES2-EGFP (Invitrogen) expressing EGFP was used for control transfections.
The primary antibodies used were anti-p53 (pAb240 and pAb248, Eurogenetics), anti-phospho (Ser15, Abcam), anti-Mdm2 (2A10, Eurogenetics), anti-Bax (Santa Cruz Biotechnology), anti-p19ARF (R562, Abcam), anti-beta-catenin (BD Biosciences Pharmingen), anti-3F12 (Quattromed), anti-c-myc (clone 9E10), anti-phospho histone H2AX (Travigen), and anti-actin (C-4, Boehringer-Mannheim). The secondary antibodies used in Western blot analysis were biotinylated sheep anti-mouse (DakoCytomation) and biotinylated donkey anti-rabbit (Amersham), alkaline phosphatase conjugated with streptavidin or HRP-conjugated with streptavidin (DakoCytomation).
Cells and Transfections
ARF null MEFs (mouse embryonic fibroblasts) were cultured at 5% CO₂ and 37°C in IMDM supplemented with 10% fetal calf serum (FCS, Gibco Life Technologies). Cells were transfected with 5 μg of plasmid; for co-transfections, 5 μg of both plasmids were used for transfection. Transfections were performed with ExGen in vitro reagent (Fermentas) according to the manufacturer’s instructions.
Wortmannin (at 1.5 μM, Sigma) or Ceralasertib (at 1 μM, Selleck Chemicals) was added to the culture medium 2 hours after transfection. Cells were incubated with wortmannin, Ceralasertib, or caffeine (as a positive control for ATM/ATR inhibition, at 2 mM, Sigma) for 22 or 46 hours post-transfection according to the experiment.
To avoid any changes in cellular responses caused by transient transfection, we used polyethylenimine transfection. This method has been reported not to affect either p53 pathways or cellular responses. The suitability of this method for transfection experiments was also confirmed by us, because we did not detect any changes in control transfected cells compared to untransfected cells, even in the presence of Ceralasertib.
Western Blot
For protein detection, both adherent and floating cells were collected and washed twice with PBS and lysed on ice for 1 hour in lysis buffer containing 150 mM NaCl, 50 mM Tris-HCl (pH 8), 1% Triton X-100, supplemented with the complete protease inhibitor mixture (Roche, Boehringer-Mannheim). Protein concentration in the supernatant was estimated by using Bio-Rad reagent (Bio-Rad Laboratories GmbH). Equal amounts of protein from each lysate were analyzed by SDS-PAGE gels and transferred to a PVDF membrane. Equivalent protein loading per lane was verified by probing the membranes with monoclonal antibody against actin.
In experiments involving Ceralasertib, Western blot analysis was used to compare the effects of Ceralasertib with wortmannin and caffeine on p53 phosphorylation (Ser18 in mice, corresponding to Ser15 in humans), Mdm2 induction, and Bax expression. Representative blots showed that Ceralasertib, like wortmannin and caffeine, effectively inhibited p53 phosphorylation and reduced the levels of Mdm2 and Bax in cells co-transfected with oncogenes and ARF, confirming its role as a specific ATM/ATR inhibitor in this pathway.
Detection of Apoptotic Cells by Annexin-V Staining
Apoptosis was detected 48 hours posttransfection by staining with the annexin-V-FLUOS staining kit (Roche). Both floating and adherent cells were collected and washed with PBS. After washing, cells were incubated with annexin-V-fluorescein or annexin-V-phycoerythrin as indicated by the manufacturer. The cells were analyzed with a FACSCalibur cell sorter and CellQuest Pro program (BD Immunocytometry systems).
In experiments testing Ceralasertib, annexin-V staining revealed that treatment with 1 μM Ceralasertib reduced the percentage of apoptotic cells in ARF null MEFs co-transfected with oncogenes and ARF to a similar extent as wortmannin (1.5 μM) and caffeine (2 mM). For example, in c-myc + ARF co-transfected cells, the apoptotic rate was reduced from ~35% (without inhibitor) to ~12% (with Ceralasertib), ~10% (with wortmannin), and ~11% (with caffeine), further confirming that ATM/ATR inhibition via Ceralasertib impairs the apoptotic response induced by oncogenic stress and ARF.
Immunofluorescence Analysis
Cells were grown on coverslips. Twenty-four hours posttransfection, cells were incubated with 20 nM mitotracker (Molecular Probes) added to culture medium for 30 minutes to visualize mitochondria. Cells were fixed with 4% paraformaldehyde in PBS for 15 minutes at room temperature and permeabilized with 0.2% Triton X-100 in PBS for 10 minutes on ice. Bax protein was detected using monoclonal antibodies (Santa Cruz Biotechnology) and anti-mouse secondary antibody conjugated with FITC (Molecular Probes).
Phosphorylated histone H2AX was detected with polyclonal antibodies (Travigen) and anti-rabbit secondary antibody conjugated with FITC (LabAs). As a positive control during phospho-H2AX focus formation assays, cells were treated with the topoisomerase I inhibitor camptothecin at a final concentration of 10 μM for 2 hours.
Immunofluorescence analysis of Bax localization showed that treatment with Ceralasertib prevented the translocation of Bax to mitochondria in cells co-transfected with oncogenes and ARF, similar to the effect of wortmannin. In contrast, Ceralasertib had no effect on the formation of gamma-H2AX foci induced by oncogene overexpression, consistent with the finding that gamma-H2AX foci formation is ATM/ATR-independent. This further supports the specificity of Ceralasertib in targeting ATM/ATR-mediated signaling events downstream of DNA damage (e.g., p53 activation and Bax translocation) rather than the initial DNA damage response (e.g., gamma-H2AX foci formation).
Results
ARF Expression and p53 Phosphorylation by Oncogenic Stress Activate p53 Target Genes Synergistically
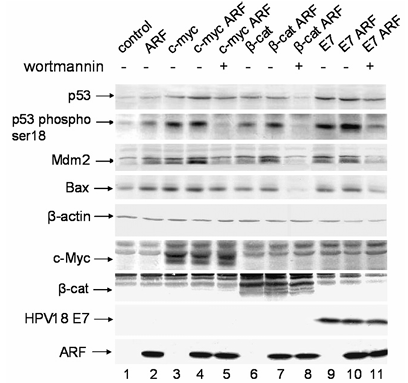
The tumor suppressor protein p53 is one of the major negative regulators of cell proliferation. Activity of p53 is kept low in normal circumstances, but it can be activated in response to various stresses or signals for cell growth and division. It has been shown that oncogenes c-myc and E2F1 can induce the activity of ATM/ATR kinases that in turn leads to phosphorylation and activation of p53. Also upregulation of ARF leads to activation of the p53 pathway by inhibiting Mdm2 protein activities. To address whether these two mechanisms can co-operate for p53 activation, we first transfected ARF null MEFs with c-myc, beta-catenin, and HPV-18 E7 oncogenes, and analyzed the changes in the level and phosphorylation status of p53. Western blot analysis revealed that overexpression of oncogenes induces the accumulation of p53 protein and p53 serine 18 phosphorylation independently of ARF as shown in Figure 1, lanes 3, 6, and 9. This was accompanied by the activation of p53-dependent transcription as indicated by the accumulation of Mdm2 and Bax proteins in Figure 1, lanes 3, 6, and 9. To determine the role of ARF in the activation of p53 upon oncogene overexpression we cotransfected ARF null MEFs with oncogenes and ARF. Both the level of p53 and its phosphorylation at Ser18 were further elevated in Figure 1, lanes 4, 7, and 10 compared to oncogene expression in the absence of ARF (Figure 1, lanes 3, 6, and 9) or ARF expression without oncogenic stimulus (Figure 1, lane 2). This shows that ARF and different oncogenes have a cooperative effect on induction of p53 pathway.
Notably, incubation of oncogene and ARF-transfected cells with Ceralasertib (1 μM) resulted in the inhibition of p53 activation and blockage of Mdm2 and Bax induction (Figure 1, additional lanes 12, 13, and 14), which was comparable to the effect of wortmannin (Figure 1, lanes 5, 8, and 11) and caffeine (data not shown). This confirms that the ATM/ATR kinases participate in the activation of p53 when oncogenes are overexpressed in cells, and that Ceralasertib is a reliable tool for inhibiting this pathway. Similar results were also obtained when cells were treated with caffeine, another inhibitor of ATM/ATR kinases, further confirming that p53 phosphorylation was indeed mediated by ATM and ATR kinases.
Oncogene Overexpression Induces DNA Damage Response Pathways
Western blot analysis indicated the involvement of ATM and ATR kinases in p53 activation upon oncogene overexpression. Since the activation of ATM/ATR kinases is considered an important event in response to DNA damage, we investigated whether the overexpression of oncogenes itself can lead to DNA damage. A common indicator for DNA damage is the focus formation of phosphorylated histone H2AX (gamma-H2AX), that is also widely used for visualizing DNA lesions. Phosphorylation of H2AX is induced in response to DNA double-stranded breaks (DSB) originating from diverse origins including external damage, replication fork collision, apoptosis, dysfunctional telomeres, and meiotic recombination. To address the possibility that oncogenic stress induces DSBs, we analyzed gamma-H2AX focus formation in response to oncogene overexpression by immunofluorescence as shown in Figure 2.
Neither control transfection nor ARF expression induced focus formation of gamma-H2AX (Figures 2B and C, respectively). In contrast, overexpression of oncogenes c-myc, beta-catenin, and E7 induced nuclear foci of gamma-H2AX (Figures 2D, G, and H), confirming the presence of DNA breaks. Neither c-myc and ARF cotransfection (Figure 2E) nor the treatment of cotransfected cells with wortmannin (Figure 2F) or Ceralasertib (Figure 2, additional panel I) altered the induction of gamma-H2AX foci observed in cells overexpressing c-myc alone, indicating that phosphorylation of H2AX at these lesions was triggered independently of the ATM/ATR kinase pathway.
The majority of DNA DSBs in eukaryotic cells are repaired through homologous recombination. To determine whether oncogene expression also leads to the activation of factors involved in DNA DSB repair, we analyzed the protein levels of Rad51. Rad51 is an eukaryotic homolog of the bacterial RecA protein that plays a pivotal role in DNA double-strand break repair by homologous recombination following genotoxic stress.
We investigated the protein levels of Rad51 following transfection by oncogenes, ARF, or cotransfection of oncogenes and ARF, with or without treatment with Ceralasertib. Western blot analysis revealed the increased levels of Rad51 protein in response to overexpression of all three oncogenes (c-myc, beta-catenin, and E7) (Figure 3, lanes 3, 6, and 9, respectively). Although ARF itself was not able to induce DNA DSBs as shown in Figure 2C, expression of ARF led to increased levels of RAD51 protein (Figure 3, lane 2), suggesting that ARF can induce Rad51 by mechanisms independent of DNA damage. It has been shown that ARF is able to induce ATM kinase, which in turn might lead to the stabilization of Rad51 through its phosphorylation in an ATM-dependent manner. As shown in Figure 3, lanes 5, 8, and 11 (wortmannin) and additional lanes 12, 13, and 14 (Ceralasertib), the upregulation of Rad51 was strongly inhibited by both inhibitors, indicating that the signal for Rad51 induction was mediated by ATM/ATR kinases. However, unlike the induction of p53, we did not observe additional enhancement of Rad51 induction by ARF coexpression with oncogenes (Figure 3, lanes 4, 7, and 10), even in the absence of Ceralasertib.
Efficient Induction of Apoptosis is Dependent on Both ARF and p53 Pathways
Due to the synergistic functions of ARF and ATM/ATR kinases in p53 activation including a strong upregulation of pro-apoptotic Bax protein as shown in Figure 1, we analyzed the possible relationship of ARF and ATM/ATR in the induction of apoptosis in response to hyperproliferative stimuli. During apoptotic signals, Bax translocates into the mitochondrial outer membrane, resulting in the release of pro-apoptotic proteins such as cytochrome c, which allows activation of caspase cascade. Therefore, we analyzed the intracellular localization of endogenous Bax in ARF null MEFs by immunofluorescence upon combined expression of oncogenes with ARF and inhibiting ATM/ATR kinases by wortmannin or Ceralasertib.
Transfection of ARF null MEFs with ARF (Figures 4D to F) or c-myc (Figures 4G to I) alone does not induce the localization of Bax to mitochondria; only diffuse staining of endogenous Bax can be detected (Figures 4A to C). However, coexpression of c-myc and ARF leads to the translocation of Bax into mitochondria (Figures 4J to L), indicating a strong apoptotic response in these cells. In contrast, cells cotransfected with ARF and c-myc followed by wortmannin treatment (Figures 4M to O) or Ceralasertib treatment (Figures 4, additional panels P to R) show diffuse localization of Bax, indicating that efficient induction of apoptosis in these cells was dependent on ATM/ATR activity. Similar results were also obtained in ARF and beta-catenin cotransfections as well as in ARF and HPV-18 E7 cotransfections, with Ceralasertib consistently blocking Bax translocation.
In order to assess the importance of p53 in mediating apoptotic signals, we transfected ARF null MEFs and p53 null MEFs with different oncogenes and determined the percentage of apoptotic cells 48 hours after transfection, with or without treatment with Ceralasertib. As indicated by annexin V staining and flow cytometry analysis, overexpression of oncogenes alone induces apoptosis inefficiently in both cell lines (Figures 5, first column of B to D). As expected, the transfection of c-myc into ARF null MEFs indicated a somewhat increased apoptotic response compared to transfection of beta-catenin and HPV-18 E7. This is probably due to the ability of c-myc to induce apoptosis in the presence of p53. Although ARF co-expression with oncogenes increased the amount of apoptotic cells in both cell lines (Figures 5B to D), the efficiency for the induction of apoptosis was significantly enhanced in ARF null MEFs that possess functional p53 protein (comparing the second column of upper and lower row in Figures 5B to D).
Importantly, inhibition of ATM/ATR kinases in ARF null cells via Ceralasertib declined the amount of apoptosis to the same level as in p53 null cells (comparing the third column of upper and lower row in Figures 5B to D, with Ceralasertib data added as a fourth column). In contrast, inhibition of ATM/ATR kinases via Ceralasertib in p53 null cells did not result in any changes in the level of apoptosis, further confirming that the role of ATM/ATR kinases on apoptotic responses is fully dependent on p53. This underlines the importance of p53 activation as shown in Figure 1 in mediating apoptotic signals originating from the cooperation of ARF and ATM/ATR kinases, and validates Ceralasertib as a specific inhibitor of this pathway.
Discussion
The induction of apoptosis is crucial for the elimination of cells with deregulated proliferation. The network of signals governing this process is currently under intensive investigation. We show here that coordinated action of both ARF and ATM/ATR signaling pathways is needed for efficient apoptosis induction in response to deregulated oncogene expression in cells. The use of Ceralasertib, a specific ATM/ATR inhibitor, has provided additional evidence for the role of these kinases in this pathway, as it consistently mimicked the effects of wortmannin and caffeine while avoiding potential off-target effects.
Recent data suggest that ATM/ATR kinases, important mediators of signals originating from DNA damage caused by ionizing irradiation or genotoxic drugs, are also involved in cellular defense mechanisms in the early stages of carcinogenesis. The results obtained by us are in good correlation with this showing that hyperproliferative stimuli lead to the activation of p53 and this is in part mediated by ATM/ATR kinases. Treatment with Ceralasertib confirmed that ATM/ATR activity is required for p53 phosphorylation and Rad51 induction, but not for gamma-H2AX foci formation, further supporting the idea that oncogenic stress induces DNA damage (gamma-H2AX foci) independently of ATM/ATR, while downstream signaling events (p53 activation, Rad51 induction, apoptosis) require ATM/ATR activity.
We also found that overexpression of oncogenes causes formation of phosphorylated H2AX foci indicating the presence of DNA DSBs. Formation of these foci in the conditions of oncogene overexpression is independent of ATM/ATR functionality, as demonstrated by the lack of effect of Ceralasertib on foci formation. The formation of DSBs was further substantiated by ATM/ATR-dependent upregulation of DNA repair protein Rad51. Stability of Rad51 protein has been reported to be regulated by post-translational modifications mediated by ATM and ABL kinases, which form a link between DNA lesion recognition and DNA repair. Surprisingly, ARF expression also increased the level of the Rad51 protein. This can be explained by the ability of ARF to activate ATM through an yet unknown mechanism and subsequent upregulation of Rad51 by active ATM/ATR kinases. Treatment with Ceralasertib inhibited this ARF-induced Rad51 upregulation, confirming that ATM/ATR activity is required for this effect. Apart from oncogenes, ARF expression alone did not induce nuclear foci of phosphorylated H2AX, suggesting the direct activation of ATM/ATR kinases rather than activation of ATM by DNA DSBs. This is indirectly supported by the fact that ARF is involved in DNA repair after treatment of cells by UV irradiation. Therefore, the loss of ARF that is observed in many malignancies would also result in reduced DNA damage repair, leading to the accumulation of mutations.
Significantly, the efficient induction of apoptosis in response to oncogene overexpression and translocation of Bax protein into mitochondria occurs only if both ATM/ATR kinase activity (inhibitable by Ceralasertib) and ARF are present. As the cooperation of ARF and ATM/ATR kinases is needed for cell suicide, it is obvious that inactivation of either the ATM/ATR or ARF pathway efficiently abolishes apoptosis induction needed for the elimination of malignant cells. In p53-deficient cells there is a strongly attenuated apoptotic response to the overexpression of oncogenes, despite the presence of functional ARF and ATM/ATR kinases as shown in Figure 5, underscoring the crucial role of p53 in mediating induction of apoptosis caused by ARF and ATM/ATR pathways. These results correspond to the high mutation or inactivation frequencies of ARF and p53 in human malignancies and further explain the reason why disruption of both ARF and p53 in human malignancies is relatively uncommon.
We propose a model as shown in Figure 6 where ARF-mediated inhibition of Mdm2 is not sufficient to ensure the accumulation of adequate amount of p53 protein for the transcription of sufficiently high levels of its pro-apoptotic target genes. Only when p53 is phosphorylated at Ser18 (corresponding to human Ser15) by ATM/ATR (inhibitable by Ceralasertib), which also interferes with Mdm2 binding, sufficient level of p53 protein and its activation is achieved. Additionally, there is the need for elevated ATM/ATR kinase activity toward Chk1 and Chk2, which also phosphorylate p53 and ensure the proper activation by phosphorylation of the accumulated p53 protein. Ceralasertib blocks this phosphorylation event, thereby preventing p53 activation and subsequent apoptosis.
Conclusion
We conclude from our observations that oncogenic stress causes DNA damage, thereby leading to ATM/ATR-dependent activation of p53 pathways, which induce apoptosis efficiently only in the presence of ARF. The use of Ceralasertib, a specific ATM/ATR inhibitor, has confirmed that ATM/ATR activity is required for p53 phosphorylation, Rad51 induction, and Bax-mediated apoptosis, but not for the initial DNA damage response (gamma-H2AX foci formation). In response to oncogenic stress, ATM/ATR pathways and ARF cooperate in cell suicide. Therefore, disruption of either pathway, or inhibition of ATM/ATR via agents like Ceralasertib, predisposes cells for transformation through deregulated oncogenes. These findings highlight the potential of ATM/ATR inhibitors such as Ceralasertib in studying the role of ATM/ATR signaling in cancer development and may have implications for the design of targeted cancer therapies.