Neuroprotection via Q-VD-OPh: Reducing Neonatal Hypoxia-Ischemia Brain Injury Through Long-Term Administration
Abstract
Apoptosis contributes greatly to the morphological and biochemical features of cell death after neonatal cerebral hypoxia-ischemia (HI), making this mode of cell death a promising therapeutic target. Previous research showed that 10 mg/kg of the caspase inhibitor Q-VD-OPh at the onset of and immediately after HI on postnatal day 9 reduced brain infarct volume. In this study, delayed administration of Q-VD-OPh, 12 and 36 hours after HI, decreased HI-induced caspase-3 activity (DEVD cleavage) by 23% and diminished the levels of the proinflammatory chemokines CCL2 (MCP-1) and CCL3 (MIP-1 alpha) by 29.3 and 29.1%, respectively, but not the levels of the anti-inflammatory cytokines IL-4 and IL-10. Long-term administration of Q-VD-OPh initiated at 12 hours after HI, and continued at 24-hour intervals for 2 weeks, reduced total brain tissue loss by 31.3% from 41.5 ± 3.1 mm3 in the vehicle group to 28.5 ± 3.0 mm3 in the Q-VD-OPh group when evaluated 16 weeks after HI (p = 0.004). Q-VD-OPh treatment also ameliorated the loss of sensorimotor function, as evaluated by a cylinder rearing test (Q-VD-OPh: 30.8 ± 4.3% vs. vehicle: 59.7 ± 6.3% in nonimpaired forepaw preference) 3 weeks after HI, and reduced HI-induced hyperactivity, as measured in an open field test (Q-VD-OPh: 4,062 ± 198 cm vs. vehicle: 4,792 ± 205 cm in distance moved) 7 weeks after the insult. However, the functional protection was no longer observed when analyzed again at later time points. The mechanisms underlying the discrepancy between sustained morphological protection and transient functional protection remain to be elucidated.
Keywords CCL2, MCP-1, CCL3, MIP-1 alpha, IL-4, IL-10, Cylinder Rearing Test, Open Field, Brain tissue loss.
Introduction
Recent studies have provided evidence that caspase activation is a prominent feature in the developing brain after hypoxia-ischemia (HI). Caspases are a family of intracellular cysteine proteases with unique substrate specificity, requiring an aspartate residue at the cleavage site, and they are essential in the execution of apoptosis. Various apoptosis-related factors, especially caspases, are upregulated in the immature brain, presumably reflecting the propensity of immature neurons to undergo discrete removal when they fail to integrate into functional networks during normal brain development, and caspase-3 has been identified as a key executor of apoptosis, the activation of which ultimately results in cell death, particularly in the immature brain. These observations support the notion that administration of caspase inhibitors is likely an effective strategy in the treatment of HI brain injury.
Several inhibitors that prevent caspase activation and apoptosis have emerged, including the widely used inhibitors carbobenzoxy-Val-Ala-Asp(OMe)-fluoromethylketone (ZVAD-FMK) and BOC-Asp(OMe)-FMK. However, when used at high doses, these inhibitors exhibit nonspecific or even toxic effects. Recently, a broad-spectrum caspase inhibitor, quinoline-Val-Asp(OMe)-CH2-PH (Q-VD-OPh) was developed. Unlike other caspase inhibitors, this has been shown to be a potent and more specific caspase inhibitor. Moreover, this compound was not toxic to cells even at high concentrations, and no obvious side effects were observed, in particular no tumor formation was seen, even following long-term (3 months) treatment.
Caspase-3-like activity is increased for at least 6 days after HI. Previously, researchers found that a single injection of Q-VD-OPh before or during HI decreased the activation of caspases, reduced infarction volume and markedly improved functional outcome after neonatal brain injury. Delayed administration, several hours after the insult, would be more relevant from a clinical perspective, but to the best of current knowledge this has not been tested for a caspase inhibitor. Moreover, new knowledge in the field of regenerative neuroscience indicates that extended treatment may have considerable beneficial effects. This led to investigation of the effects of delayed and extended Q-VD-OPh administration on the immature brain after HI.
Materials and Methods
Ethics Statement
All animal experiments were approved by the local Animal Ethics Committee at the University of Gothenburg (ethical approval No. 90-2011).
Animals
Postnatal day (PND) 8 C57BL/6 male mice were purchased from Charles River Laboratories (Sulzfeld, Germany). The mice were housed in a 12-hour light/dark cycle. Standard laboratory chow (B&K, Solna, Sweden) and drinking water were available ad libitum.
Hypoxia-Ischemia
The mice were subjected to unilateral HI on PND 9 according to the Vannucci model adapted for mice. Briefly, the mice were anesthetized with isoflurane (5.0% for induction and 1.5-3.0% for maintenance) in a mixture of nitrous oxide and oxygen (1:1). The left common carotid artery of each pup was exposed, isolated from the nerve and vein, and ligated with Prolene sutures (6.0). After the wound was sutured the mice were returned to their dams, allowed to recover for 1 hour, and then placed in an incubator perfused with a humidified gas mixture (10% oxygen in nitrogen) at 36 degrees Celsius for 50 minutes. Control animals did not undergo surgery and hypoxia.
Q-VD-OPh Administration
The broad-spectrum caspase inhibitor Q-VD-OPh (SM Biochemicals, Anaheim, California, USA) was dissolved in 100% DMSO and further diluted with saline to produce a 1 mg/ml working solution in 10% DMSO. The working solution was initially injected intraperitoneally at a dose of 10 mg/kg 12 hours after the HI insult. For caspase-3-like activity and Luminex assays, the second injection followed 36 hours after HI. For long-term evaluation, additional injections were administrated at 24-hour intervals for a total period of 14 days. Control pups received an equivalent volume of saline containing 10% DMSO.
Sample Preparation for Caspase Activity and Luminex Assays
The animals were sacrificed by decapitation at 48 hours after HI. Tissue from the parietal cortex (including hippocampus) in each hemisphere were rapidly dissected out on a bed of ice and snap frozen in dry ice. Tissue samples were homogenized by sonication in 1,000 microliters ice-cold 50 mM Tris-HCl (pH 7.3) solution containing 5 mM EDTA, and then aliquoted and stored at -80 degrees Celsius.
Caspase-3-Like Activity Assay
Protein concentration was determined using a bicinchoninic acid protein assay kit (Thermo, Stockholm, Sweden). Samples of homogenate (25 microliters) were mixed with 75 microliters extraction buffer containing 50 mM Tris-HCl (pH 7.3), 100 mM NaCl, 5 mM EDTA, 1 mM EGTA, 3 mM NaN3, 1 mM PMSF, 0.5% protease inhibitor cocktail and 0.2% CHAPS (all products are from Sigma, Stockholm, Sweden) on a microtiter plate (Thermo). After incubation for 15 minutes at room temperature, 50 micromolar Ac-Asp-Glu-Val-Asp-aminomethyl coumarine (Ac-DEVD-AMC; VWR, Stockholm, Sweden) in 100 microliters extraction buffer, without inhibitors or CHAPS but with 4 mM DTT, was added. Cleavage of the substrate was measured at room temperature every 2 minutes for 30 minutes using a microplate fluorescence reader (FLUOstar Omega, BMG Labtech, Germany) at an excitation wavelength of 355 nm and an emission wavelength of 460 nm. The cleavage activity was obtained from the slope by plotting fluorescence units against time. A standard curve with AMC concentrations ranging from 0-100 micromolar in appropriate buffer was used to express the data in picomoles of AMC formed per minute per milligram of protein.
Luminex Assay
Homogenate samples used for analysis of brain cytokines and chemokines were centrifuged at 10,000 g for 10 minutes at 4 degrees Celsius to obtain crude cytosolic fractions and stored at -80 degrees Celsius. Total protein concentration was measured as described above. Two proinflammatory chemokines, CCL2 and CCL3 (also called MCP-1 and MIP-1 alpha, respectively) and two anti-inflammatory cytokines, IL-4 and IL-10, were assayed using a custom-designed 4-plex mouse cytokine assay kit (Bio-Rad, Stockholm, Sweden). The samples were centrifuged at 10,000 g for 10 minutes to remove debris prior to analysis. The assay was performed according to the protocol provided by the manufacturer with some modifications; the protein concentrations for the samples used for analysis were higher than 2 mg/ml, and 0.5% BSA was added prior to analysis to minimize protein loss in the system. Results are expressed as picogram/milligram protein.
Cylinder Rearing Test
A cylinder rearing test was used to evaluate forepaw use asymmetry, as described previously. Briefly, each animal was placed in a transparent glass cylinder (95 mm diameter, 180 mm height) and video-recorded. The initial forepaw (left/right/both) preference during full rearing was monitored for 3 minutes at 3 and 12 weeks after HI. Forepaw use at the first contact with the cylinder wall during full rearing was recorded as left (nonimpaired), right (impaired) or both, where both equals no preference. The proportion of left forepaw preference was calculated as follows: (left forepaw preference minus right forepaw preference)/(left forepaw preference plus right forepaw preference plus both) times 100%.
Open Field
The motor activity patterns were analyzed by using open field and video tracking as previously described. Each animal was introduced to an unfamiliar, open field arena and then immediately videotaped at a sampling frequency of 12.5 Hz for 10 minutes at 7 and 15 weeks after HI. Four indirectly illuminated arenas were videotaped simultaneously. The floor of each arena was covered with gray gravel previously exposed to other mice. Recorded videos were analyzed using Viewer 6.1 (BIOBSERVE GmbH, St. Augustin, Germany). Data analyzed include distance moved, number of stops and time spent in the middle of the arena.
Immunohistochemistry
Animals were deeply anesthetized with 50 mg/ml phenobarbital and perfused intracardially with 4% paraformaldehyde in 0.1 M phosphate buffer solution through the ascending aorta for 5 minutes. The brains were immediately removed and postfixed at 4 degrees Celsius for 24 hours. The brains were dehydrated with graded ethanol and xylene, paraffin-embedded, and cut into 5 micrometer-thick coronal sections. Every 100th section for MAP-2 staining and every 50th section for Iba-1 staining were deparaffinized in xylene and rehydrated in graded ethanol concentrations. Antigen retrieval was performed by heating the slides in 10 mM sodium citrate buffer, pH 6.0, for 10 minutes. Nonspecific binding was blocked with 4% horse serum in phosphate-buffered saline (PBS) for 30 minutes. Sections were incubated for 60 minutes with primary antibodies diluted in PBS (1:1,000 mouse anti-MAP-2, clone HM-2; Sigma; 1:1,000 rabbit anti-Iba1, WAKO, Osaka, Japan) followed by 60 minutes incubation with biotinylated secondary antibodies in PBS (1:200; the secondary antibodies were from Jackson ImmunoResearch Laboratories, Stockholm, Sweden). After blocking endogenous peroxidase activity with 3% H2O2, the sections were visualized with Vector ABC Elite and 3,3-diaminobenzidine (DAB), and mounted using Vector mounting medium. For double staining of Iba-1 and galectin-3, mixed primary antibodies (1:500 rabbit anti-Iba-1; Wako and 1:100 rat anti-galectin-3; eBioscience) were incubated for 60 minutes at room temperature following antigen retrieval. After washing, the sections were incubated with fluorophore-conjugated secondary antibodies (1:1,000 Alexa Fluor 488 donkey anti-rat IgG (H+L) and 1:1,000 Alexa 555 donkey anti-rabbit IgG (H+L); Invitrogen). After washing, the sections were mounted using Prolong antifade mounting medium.
Injury Evaluation
The MAP-2 stained areas were measured in both hemispheres using Micro Image (Olympus, Japan). The volume of tissue loss was defined as the MAP-2-positive volume in the contralateral hemisphere minus that of the ipsilateral hemisphere, and was calculated according to the Cavalieri principle using the following formula: V = sum A times P times T, where V is the total volume, sum A is the sum of the areas measured, P is the inverse of the sampling fraction and T is the section thickness. The neuropathology score was evaluated in MAP-2-stained sections using a semiquantitative scoring system, as described previously.
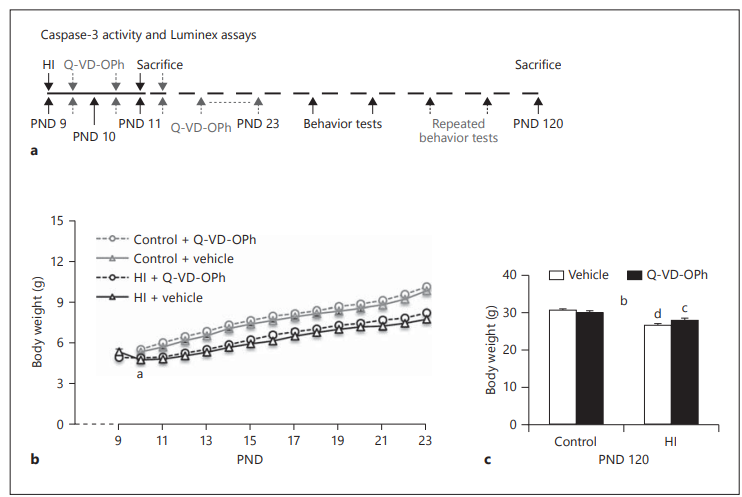
Figure 1: Body Weights Overview
Panel (a): Study Design
- Depicts the experimental timeline, likely showing:
- Induction of hypoxia-ischemia (HI)
- Treatment windows (Vehicle vs. Q-VD-OPh)
- Time points for body weight measurements
- Final endpoint at PND 120 (Postnatal Day 120)
Panel (b): Body Weights Over Time (0–24 h post-HI)
- HI mice had significantly lower body weights compared to non-HI (control) littermates during the first 2 weeks, regardless of treatment.
- Statistical significance:
- a p<0.01a \, p < 0.01ap<0.01: Repeated measures test shows significant drop in body weight for vehicle-treated HI mice between 0 and 24 hours post-HI.
Panel (c): Body Weights at PND 120
- Final weight measurements prior to experimental endpoint.
- Statistical findings:
- b p<0.05b \, p < 0.05bp<0.05: Significant interaction between treatment and brain injury (Two-way ANOVA).
- c p<0.05c \, p < 0.05cp<0.05, d p<0.001d \, p < 0.001dp<0.001: Significant differences between experimental groups compared to corresponding control groups.
Cell Counting
Iba-1-positive cells were counted using stereological principles in the peri-infarct regions (Stereo Investigator; MicroBrightField, Colchester, Vermont, USA). The peri-infarct region was defined as an area with a radius of 300 micrometers adjacent to the edge of the infarct cavities (after 4 months maturation). For normal controls, 3 visual fields (1 visual field = 0.784 mm2) were randomly chosen in the corresponding brain region, and cells were counted within each section. Immunopositive cells were counted in every 50th section, with an analysis of 3-4 sections per animal. The total number of cells was obtained by multiplying the number of cells by the sampling fractions. Results are reported as number of positive cells per mm3.
Statistics
An independent Student’s t-test was used when comparing injury scores and tissue loss. A repeated measures test was used to compare the body weight gain over the 2-week injection period. A two-way ANOVA was used when two factors were involved, such as a treatment and brain injury. Treatment, brain injury and an interaction between the two were considered as main effects. When significant interaction was detected, one-way ANOVA was used to analyze the effects of treatment and injury separately. Statistical analysis was performed using SPSS 21.0 (SPSS, IBM, New York, NY, USA) and significance was assumed when p < 0.05. All data are shown as means ± SEM.
Results
Body Weight Gain
Body weight gain is an easy, sensitive approach for health monitoring in young rodents. In this study, animals were weighed daily for 14 days, concurrent with the daily intraperitoneal injections, and on PND 120 before sacrifice to observe their general health status. Vehicle-treated ischemic mouse pups (n = 19) showed a significant reduction in body weight 24 hours after HI (PND 10) when compared with their body weights immediately after the HI insult (4.75 ± 0.21 g at 24 hours after ischemia vs. 5.27 ± 0.16 g at 0 hours after ischemia, p < 0.01), whereas the body weight of Q-VD-OPh-treated mice (n = 18) was stable up to 24 hours after HI (4.85 ± 0.14 vs. 4.82 ± 0.13 g). All mice subjected to HI, regardless of treatment, recovered and resumed growth 48 hours after ischemia, but they did not catch up with their non-HI littermates during the 2 weeks of vehicle or Q-VD-OPh treatment (control + vehicle, n = 20; control + Q-VD-OPh, n = 19; Figure 1b). Q-VD-OPh per se did not affect the overall growth; no significant difference was observed in body weight gain between vehicle-treated and Q-VD-OPh-treated mice at any of the measured time points from PND 11 to PND 23 (Figure 1b). By PND 120, two-way ANOVA revealed a significant interaction between treatment and brain injury in body weight (F(1,72) = 5.14, p < 0.05). Although the Q-VD-OPh-treated mice displayed a closer-to-normal body weight after HI (28.08 ± 0.49 g in Q-VD-OPh-treated ischemic mice vs. 26.62 ± 0.46 g in vehicle-treated ischemic mice), the body weights did not differ according to treatment (Q-VD-OPh) and the body weights of all HI mice were still lower than those of their non-HI littermates (Figure 1c).
Q-VD-OPh Decreased Caspase-3 Activation and Diminished the Levels of Proinflammatory Chemokines, but Not Anti-Inflammatory Cytokines
The animals received the first Q-VD-OPh administration as late as 12 hours after the HI insult (n = 8) but nevertheless displayed significantly lower caspase-3-like activity 48 hours after HI compared with vehicle-treated ischemic littermates (n = 7; Q-VD-OPh 177.4 ± 13.5 vs. vehicle 229.7 ± 7.3 pmol AMC per minute per milligram protein, p = 0.006; Figure 2a).
No expression of CCL2 and very low expression of CCL3 were detected in the brains of normal mice (control + vehicle, n = 8; control + Q-VD-OPh, n = 7). At 48 hours after HI, the levels of these two chemokines increased dramatically. Compared with vehicle-treated ischemic mice, animals that received Q-VD-OPh at 12 and 36 hours after HI had significantly lower levels of CCL2 (Q-VD-OPh 27.4 ± 2.7 vs. vehicle 38.7 ± 3.4 pg/mg protein, p < 0.05) and CCL3 (Q-VD-OPh 35.6 ± 1.8 vs. vehicle 50.2 ± 6.6 pg/mg protein, p < 0.05; Figure 2b, c). The anti-inflammatory cytokines, IL-4 and IL-10, were expressed in normal brains, and their expression levels increased after ischemic injury, but Q-VD-OPh did not diminish the increase (Figure 2d, e).
Q-VD-OPh Did Not Change Microglia Densities 4 Months after HI
Iba-1 is a general marker for microglia and galectin-3 is a marker of pathologically activated microglia. In the current study, no galectin-3-positive cells were observed in the injured brains 4 months after the insult. The number of Iba-1-positive cells was quantified in the peri-infarct area, as described in Methods. Long-term treatment with Q-VD-OPh did not change microglia densities in Q-VD-OPh-treated ischemic mice, neither compared with vehicle-treated mice nor with normal controls. Furthermore, at this late time point, 4 months after HI, all the microglia displayed a ramified, surveillance morphology. There were no amoeboid, activated microglia in the peri-infarct area.
Q-VD-OPh Reduced Tissue Loss after HI
On PND 120, the mice were sacrificed for brain injury evaluation. Long-term administration of Q-VD-OPh significantly reduced tissue loss by 31.3% compared with the vehicle-treated ischemic animals (Figure 3a). The tissue loss in the vehicle group was 41.5 ± 3.1 mm3 (n = 19) versus 28.5 ± 3.0 mm3 (n = 18) in the Q-VD-OPh group at 16 weeks after the HI insult (p = 0.004; Figure 3b). Neuropathological scores were significantly lower in the cortex, hippocampus and thalamus (but not the striatum) compared with the vehicle-treated controls (Figure 3c).
Q-VD-OPh Improved Motor Function Early, but Not Late after HI
To determine whether Q-VD-OPh treatment ameliorated functional deficits, a cylinder rearing test was performed 3 weeks and 12 weeks post-HI. In the cylinder rearing test performed 3 weeks after HI, normal control animals used either their left or right forepaw evenly when touching the cylinder wall during full rearing. Two-way ANOVA analysis indicated a significant interaction between treatment and brain injury (F(1,72) = 16.39, p < 0.001). The vehicle-treated ischemic mice (n = 19) showed a 60% increase in left forepaw preference, in line with left hemisphere injury, indicating a sensorimotor deficit and impaired use of the right forepaw (p < 0.001; Figure 4a). Q-VD-OPh treatment (n = 18) significantly improved sensorimotor performance (p < 0.001; Figure 4a). Surprisingly, results from the cylinder rearing test 12 weeks after HI showed an increase in left forepaw preference both in vehicle-treated HI mice and in Q-VD-OPh-treated HI mice and the amelioration conferred by Q-VD-OPh was no longer observed (Figure 4b).
Q-VD-OPh Normalized Motor Hyperactivity Early, but Not Late after HI
An open field test was performed at 7 and 15 weeks post-HI. A significant interaction between treatment and brain injury was revealed by two-way ANOVA analysis (F(1,72) = 4.54, p < 0.05). The total distance moved during the open field test 7 weeks after HI revealed that the vehicle-treated HI mice displayed an increased motor activity compared to non-HI controls (p < 0.001; Figure 4c). Q-VD-OPh treatment normalized the HI-induced hyperactivity (Q-VD-OPh 4,062 ± 198 cm, n = 18, vs. vehicle 4,792 ± 205 cm, n = 19, p < 0.05; Figure 4c). At 15 weeks after HI, all animals moved less than at 7 weeks after HI in the same arena. However, at this later time point, Q-VD-OPh treatment did not normalize the hyperactivity (Figure 4d); both vehicle-treated and Q-VD-OPh-treated ischemic mice displayed a similar, approximately 50%, increase in the distance moved when compared with controls (Figure 4d). There were no differences in the other two measured parameters (number of stops or time spent in the middle of the arena), either between vehicle-treated controls and vehicle-treated HI mice or between vehicle-treated HI mice and Q-VD-OPh-treated HI mice.
Discussion
In the present study, the effects of delayed and extended systemic Q-VD-OPh administration on postischemic inflammation, morphological brain damage and functional deficits induced by neonatal HI were examined. Q-VD-OPh injections initiated as late as 12 hours after the insult and supplemented with an additional injection 36 hours after HI decreased the expression of the proinflammatory chemokines CCL2 (MCP-1) and CCL3 (MIP-1 alpha), but not the anti-inflammatory cytokines IL-4 and IL-10. Long-term administration of Q-VD-OPh (daily injections for a total period of 2 weeks starting from 12 hours post-HI) reduced brain injury and ameliorated HI-induced sensorimotor deficits and hyperactivity. However, this amelioration of functional deficits was not long-lasting.
Postischemic Caspase Activity and Inflammation
Caspase-3 is the most abundant effector caspase and plays a central role in apoptotic cell death in the immature brain, displaying increased activity for at least 6 days after HI. It is well accepted that caspase activation is more important in the immature brain for the development of injury. In addition, in vitro and in vivo studies have demonstrated sexual dimorphism regarding apoptosis-related mechanisms, with male neurons displaying more pronounced translocation of apoptosis-inducing factor (caspase-independent apoptosis) and female neurons a stronger activation of caspase-3. Only male mice were used in this study, but it is conceivable that more robust neuroprotection would be achieved with caspase inhibition in females. Cytokine production by resident brain cells, mainly microglia, has been demonstrated. Some of them, like the chemokines CCL2 and CCL3, are involved in guiding monocytes/macrophages toward the ischemic area and appear to exacerbate cerebral injury. Others, like IL-4 and IL-10, are cytokines with anti-inflammatory properties, and thus exert protective or regenerative mechanisms after ischemic brain injury. In this study, both the caspase-3-like activity and expression levels of 4 different chemokines/cytokines were assayed 48 hours after ischemia and the number of microglia was quantified 4 months after the insult. Two injections of Q-VD-OPh reduced caspase-3-like activity by 23% and decreased the expression of CCL2 and CCL3 by 29.3 and 29.1%, respectively, but not the levels of IL-4 and IL-10. These results are in accordance with earlier findings, demonstrating less brain injury after caspase inhibition in rodent models of injury to the immature brain. However, the role of Q-VD-OPh in postischemic inflammation is unclear. In the light of recent findings demonstrating that microglia activation requires caspase activation, albeit at lower levels than in apoptotic cell death, it is tempting to speculate that part of the protective effect observed is due to reduced inflammation, particularly since the anti-inflammatory cytokines IL-4 and IL-10 were unaffected. It remains to be shown if caspase inhibition in microglia specifically prevents the formation of the toxic so-called M1 phenotype, but not the regenerative so-called M2 phenotype. However, the lower levels of proinflammatory chemokines may also simply be secondary to the lesser injury. To investigate this would require detailed time courses of cytokine expression levels and caspase-3 activity in the presence or absence of caspase inhibition, but this is beyond the scope of the present study.
Discrepancy between Morphological and Functional Protection
The current study differs from previous reports of Q-VD-OPh treatment in ischemia-induced brain injury in that long-lasting neurobehavioral outcomes were examined. After PND 60, when brain development is completed, the two functional tests were repeated. Disappointingly, the functional improvements conferred by Q-VD-OPh earlier were no longer observable. The mechanisms underlying the discrepancy between the morphological, long-term protection and the transient functional protection remain to be elucidated. Magnetic resonance imaging studies in human infants show that neurodegeneration in multiple integrated brain regions evolves over time after the initial HI insult. Moreover, previous research has shown that transplantation of mesenchymal stem cells markedly ameliorated sensorimotor deficits and decreased brain tissue loss in a mouse model of HI, but analogous to current findings functional impairment still increased over time. Thus, even though Q-VD-OPh reduced tissue loss in the current study, it likely just postponed neural functional loss after HI.
Benefit of Acute versus Chronic Caspase Inhibition after Injury to the Immature Brain
Previous data showed that injection of Q-VD-OPh at 10 mg/kg administered at the onset of and immediately after HI reduced the infarct volume by 48% as quantified 3 days after the insult. An advantage with Q-VD-OPh is low toxicity. Previous research treated adult mice with 10 mg/kg Q-VD-OPh (3 injections per week for 3 months) without observing any apparent adverse effects. In the current study delayed and long-term administration was explored, demonstrating 31% protection 4 months after HI after 14 injections of Q-VD-OPh. The protection was less impressive than the 48% obtained after only two injections of Q-VD-OPh, the most likely reason being the delayed onset of treatment. Much of the caspase activation after HI had already happened 12 hours after the insult; this is supported by the 57% decrease in caspase-3-like activity detected 24 hours after the insult in previous work, while in the current study only a 23% decrease was observed 48 hours after the injury. In addition, the interval between HI and evaluation of brain injury was also very different (3 days vs. 4 months) and it remains to be shown what the tissue protection would be 4 months after HI and with only two injections of Q-VD-OPh.
In summary, delayed and extended administration of Q-VD-OPh decreased postischemic inflammation, reduced brain damage and transiently ameliorated functional deficits induced by neonatal HI. Further investigations are necessary to clarify the role of caspase inhibitors in postischemic inflammation and the mechanisms underlying the discrepancy between sustained morphological protection and transient functional protection.